
Abstract
Bioreactors are essential cell and tissue culture tools that allow the introduction of biophysical signals into in vitro cultures. One major limitation is the need to interrupt experiments and sacrifice samples at certain time points for analyses. To address this issue, we designed a bioreactor that combines high-resolution contact-free imaging and continuous flow in a closed system that is compatible with various types of microscopes. The high throughput fluid flow bioreactor was combined with two-photon fluorescence lifetime imaging microscopy (2P-FLIM) and validated. The hydrodynamics of the bioreactor chamber were characterized using COMSOL. The simulation of shear stress indicated that the bioreactor system provides homogeneous and reproducible flow conditions. The designed bioreactor was used to investigate the effects of low shear stress on human umbilical vein endothelial cells (HUVECs). In a scratch assay, we observed decreased migration of HUVECs under shear stress conditions. Furthermore, metabolic activity shifts from glycolysis to oxidative phosphorylation-dependent mechanisms in HUVECs cultured under low shear stress conditions were detected using 2P-FLIM. Future applications for this bioreactor range from observing cell fate development in real-time to monitoring the environmental effects on cells or metabolic changes due to drug applications.
Export citation and abstract BibTeX RIS

Original content from this work may be used under the terms of the Creative Commons Attribution 3.0 licence. Any further distribution of this work must maintain attribution to the author(s) and the title of the work, journal citation and DOI.
Introduction
Fluid shear stress, due to blood flow through vessels, plays a crucial role in angiogenesis [1], cell proliferation [2], migration [3], differentiation [2, 4], and metabolism [5]. Cells are sensitive to shear stress, which can ultimately alter cell function through activation of mechanosensitive molecules involved in cell signaling pathways [6]. Bioreactor systems are commonly closed systems that employ mechanical stimuli to cells and in vitro-generated cell-based three-dimensional (3D) tissues with the goal to mimic in vivo physiological conditions [7]. Depending on the type of cell or tissue, parameters such as temperature, oxygen concentration, mechanical and electrical stimulation [8] are the typically manipulated and monitored in bioreactor systems. One limitation of the majority of the currently existing bioreactor systems is the need to interrupt the culture at defined time points to gather information about the status of the sample using invasive methods. High-resolution optical techniques such as Raman microspectroscopy and near-infrared two-photon fluorescence lifetime imaging microscopy (2P-FLIM) are widely used to detect cell phenotypic changes, metabolic activity, and protein expression of cells without the need for exogenous markers [9–11]. It would be highly advantageous to develop a bioreactor system that allows real-time high-resolution imaging of cells and tissues that are exposed to biomechanical stress in vitro without the need to stop the experiment or sacrifice the sample. However, to combine high-resolution imaging modalities with continuous flow bioreactor systems is challenging due to short working distances and constraints of optical transparency. Although some custom-built and commercially available devices have been developed to apply shear stress in vitro, few existing designs are optimized to allow for simultaneous high-resolution live-cell microscopy [4, 12, 13]. Microfluidic devices were designed for high-resolution live-cell microscopy. However, these devices also face technical challenges [14]. For example, bubbles in the microfluidic channels/chambers are difficult to remove and may damage cells, therefore hindering the control of these devices [14]. Furthermore, unlike bioreactor systems, microfluidic devices are designed to culture and analyze single cells or monolayers of cells, but they are not suitable for the culture of larger samples such as 3D tissues [14].
Here, we present a bioreactor that combines high-resolution contact-free imaging and continuous flow in a closed system that is compatible with various types of microscopes. 2P-FLIM has recently emerged in the field biology as a tool to study complex biological samples [15–17]. It is a non-invasive method that relies on the principle that intrinsic molecules within cells, such as amino acids, proteins, and lipids often emit a well-perceivable fluorescence after excitation with light in the ultraviolet or visible range [18]. This excitation is accomplished using near-infrared femtosecond multiphoton lasers. Therefore, no exogenous fluorescent dyes must be added to the culture for visualization and analysis. In addition to cellular and tissue autofluorescence, 2P-FLIM can detect pH level, cation (Ca2+, Mg+, K+) concentration, oxygen concentration and fluorescence resonant energy transfer [19–23]. NAD(P)H (nicotinamide adenine dinucleotide (phosphate)) is a key enzyme involved in glycolysis and oxidative phosphorylation [11, 23, 24]. Changes of NAD(P)H and FAD (flavin adenine dinucleotide) are reflective of cellular metabolism [25] that can be non-invasively detected with 2P-FLIM. Shear stress has been shown to influence metabolic activity and migration of endothelial cells [26], therefore, we aimed to investigate the influence of shear stress on cell migration, cell metabolism and apoptosis of human umbilical vein endothelial cells (HUVECs) by implementing bright field imaging and 2P-FLIM in our bioreactor system.
Materials and methods
Cell culture
HUVECs were obtained from Lonza (CC-2517, Basal, Switzerland). Passages 3–6 were used for all experiments with the corresponding EBM-2 MV BulletKit medium (CC-3156 and CC-4147; Lonza).
For the bioreactor experiments, HUVECs were trypsinized (0.25% trypsin-EDTA) and re-seeded at a density of 3 × 104 cells cm−2 on circular 25 mm diameter coverslips (Nunc Thermanox, Thermo Fischer Scientific, Langselbold, Germany). The cell-seeded coverslips were cultured overnight in a six-well plate at 37 °C and 5% CO2 to allow cell attachment prior transfer into the bioreactor system.
Imaging bioreactor system
A custom-built, autoclavable imaging bioreactor system was designed using Solidworks (Solidworks2010, Dassault Systemes SolidWorks Corporation, Ludwigsburg, Germany) (figure 1). A computational model was developed to assess the performance of the bioreactor system in silico. Meshing and calculations were performed in COMSOL 4.3a based on the bioreactor geometry and dimensions (COMSOL Multiphysics GmbH, Berlin, Germany). The medium was considered as an incompressive Newtonian fluid. Therefore, the equation used in the simulation was incompressive Navier–Stokes equations as follows (equations (1) and (2)):
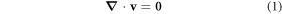
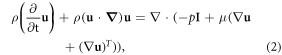
where u is the fluid velocity, ρ is the fluid density, p is the fluid pressure, μ is the fluid dynamic viscosity of the culture medium and F is the volume forces vector. The numerical values of the model parameters used in the simulations are displayed in table 1.

Figure 1. Overview and in silico characterization of the imaging bioreactor employed in this study. (a) SolidWorks computer-aided design drawing. Cross-section of bioreactor flow chamber with the white arrows indicating the flow of medium, the top component is blue and the yellow color depicts the cell-seeded area. (b) Bottom view of the bioreactor system shows the four distinct flow chambers. (c) Bioreactor in the experimental set up: fluorescence microscope with built-in incubator for live-cell imaging. (d) COMSOL in silico characterization of flow dynamics using a flow rate of 1.48 ml min−1. Red stream lines show laminar flow through the chamber. The applied shear stress on the cell-seeded substrate is visualized via a color code.
Download figure:
Standard image High-resolution imageTable 1. Numerical values of the model parameters used for the simulation.
| Parameters (units) | Value | Reference |
|---|---|---|
| Inlet velocity | 0 | |
| Outlet pressure (Pa) | 0 | |
| Boundaries | No slip | |
| Fluid viscosity (Pa s) | 8.1 × 10−4 | [51, 52] |
| Fluid density at 37 °C | 103 | [51, 52] |
| Temperature (K) | 310.15 | [51, 52] |
The bioreactor system consists of three parts: a transparent chamber top (made of polycarbonate), a flow chamber block and a bottom (both made of poly-ether-ether-ketone) (figure 1(a)). For all experiments, cell-seeded coverslips were placed between the flow chamber block and bottom. There are four fluidic chambers; each with a distinct inlet and outlet valve to enable laminar flow. Each fluidic chamber was connected to a closed tubing system (Ismatec, Wertheim-Mondfeld, Germany) containing a 30 ml medium reservoir (Schott AG, Mainz, Germany) and a filter with a 20 μm pore size (Whatman GmbH, Dassel, Germany). The cell medium was pumped from the reservoir through the system using a peristaltic pump with a CA8 pump head and cassette (Ismatec). All in vitro experiments were performed at 37 °C and 5% CO2.
Live-cell bright field microscopy
After 24 h of static cell culture, a p100 pipette tip (Eppendorf, Hamburg, Germany) was used to create a scratch and then coverslips were placed in the bioreactor system, with the scratch perpendicular to flow direction, medium was added, the closed system was connected to the medium pump and placed on the microscope stage for live-cell and 2P-FLIM data acquisition.
In this study, we utilized a Zeis AxioObserver Z1 (Carl Zeiss GmbH, Jena, Germany) with live-cell imaging compatibility and an attached, closed incubator to ensure 37 °C and 5% CO2 during the entire experiment (figure 1(c)). A bright-field imaging channel and a 40× water immersion objective (N.A 1.1, Carl Zeiss GmbH) were used to acquire images every 5 min for 20 h after initial injury. The focus was set and fixed prior to the experiment.
Immunofluorescence and live/dead cell staining
After 24 h of culture within the bioreactor system, the cell-seeded coverslips were extracted from the bioreactor, fixed with 4% paraformaldehyde for 15 min, washed and then stained for the endothelial cell marker CD31 as described previously in detail [24]. Cell nuclei were visualized using 4', 6-Diamidin-2-phenylindol (DAPI). Mouse IgG1 PECAM 1 (CD31; sc-71872, 1:100; Santa Cruz, Heidelberg, Germany) served as the primary antibody, and anti-mouse-IgG1-Alexa Fluor 594 (A-11005, 1:250; Life Technologies, Carlsbad, USA) was used as the secondary antibody. A confocal microscope LSM710 (Carl Zeiss GmbH) was used to obtain the images, which were processed using Photoshop CS3 (Adobe Systems, San Jose, USA).
To further characterize the bioreactor system, cell viability following a 24 h dynamic culture was assessed using a live/dead staining protocol. Fluorescein diacetate (FDA; Sigma-Aldrich, Darmstadt, Germany) at a concentration of 1 μg ml−1 and propidium iodide (PI; Sigma-Aldrich) at a concentration of 1 μg ml−1 were used. Dilutions were prepared with cell culture medium without fetal calf serum. For staining, the cells were washed with PBS and incubated for 15 min in the dark at 37 °C with the dye solution. The dye was then removed, the cells were washed with PBS and images were immediately acquired.
Two-photon fluorescence lifetime imaging microscopy (2P-FLIM)
2P-FLIM measurements were performed on a custom-made multiphoton laser system (JenLab GmbH, Jena, Germany) that has been previously described in detail [11, 25]. Two-photon excitation was generated using a Ti:Sapphire femtosecond laser (MaiTai XF1 Spectra Physics, United States, Santa Clara). The following settings were maintained for all 2P-FLIM experiments: multiphoton images were collected in the region of interest with a total acquisition time of 23 s, a laser power of 25 mW and a wavelength of 710 nm. Afterwards, the microscope was switched to 2P-FLIM mode for the acquisition of the decay data. All recording settings were adjusted using the software SPCM (Becker & Hickl GmbH, Berlin, Germany). Each 2P-FLIM measurement had a total recording time of 180 s and was controlled through the SPCM software (Becker & Hickl GmbH). Instrumental response function (IRF) was considered to acquire accurate results [26, 27]. The measurement of IRF was performed as previously described [28]. In detail, amorphous urea (Sigma-Aldrich) was dissolved in distilled water and added to a glass bottom dish (ibidi GmbH, Martinsried, Germany). After resting overnight, the water was evaporated, and the saturated concentration of urea formed crystals on the glass surface. Urea crystals were then measured to acquire the time-resolved scattering signal coming from the crystals. The excitation wavelength was 920 nm and acquisition time was 180 s with a laser power of 5 mW.
2P-FLIM data analysis
It is known that fluorescence signals originating from NAD(P)H contain contributions from free and protein-bound NAD(P)H [29]. Biexponential decay fitting was used to calculate the short and long lifetime components (τ1 and τ2, respectively), which mostly correspond to free and protein-bound NAD(P)H [30]. The relative contributions of the lifetime components (α1 and α2, where α1 + α2 = 100%) were also calculated. The IRF was recorded from urea crystals, and the instrumental delays were taken into account in the lifetime calculations as previously described [11]. At the end of every fitting, χ2, a parameter expressing the quality and accuracy of the biexponential decay fitting, was calculated. The fitting parameters were reevaluated until the average χ2 per image was lower than 1.1. Every pixel was independently calculated to enable false color-coding based on the values of the different parameters (τ, α, and χ2). Afterwards, every decay map pixel (2P-FLIM image) was exported as a text file and analyzed using ImageJ (freely available from www.nih.org). All data analyses were performed with the software SPCM (Becker & Hickl GmbH) with a binning factor of 6 and a threshold of approximately 30% of the maximum signal.
Statistical analysis
Normal data distribution was assessed using a Shapiro–Wilk test. For statistical analysis, the commercially available program GraphPad Prism 6 was used (GraphPad Software, Inc., La Jolla, CA). Students t-test was performed, and p < 0.05 was considered statistically significant.
Results
The imaging bioreactor system was designed and fabricated to allow cell and tissue cultures to be exposed to defined fluid shear stress while enabling high-resolution imaging (figure 1(a)). It has the same dimension as a typical cell culture plate (127 mm × 85 mm × 21 mm) making it compatible with various existing microscope systems. The bioreactor has four distinct flow chambers with no communication or connection between the chambers (figure 1(b)), enabling the execution of four independent experiments simultaneously. The transparent top component allows bright field light penetration and is suitable for inverted light microscopes. Windows were designed in the bottom component to ensure contact between the imaging objective and the cell-seeded area within the bioreactor system to allow for high-resolution imaging (figure 1(c)). In our proof-of-concept experiment, cell-seeded coverslips were placed between the chamber block and bottom, with an O-ring seal. The coverslips are removable from the bioreactor system for end-point biological analysis. To characterize the dynamics of the flow chamber, simulations were performed using a computational simulation COMSOL. We simulated steady/state flow patterns within the fluidic chamber under the assumption of a fully-developed flow. As shown in figure 1(d), a constant shear stress between 1.69 × 10−4 and 4.36 × 10−3 dyn cm−2 was applied over the entire cell-seeded coverslip with a flow rate of 1.48 ml min−1. The red streamlines confirmed a laminar flow of medium through the chamber. We identified 47 ml s−1 as the maximum flow rate to ensure laminar flow to the cells (figure 1(e)). With this flow rate, cell-seeded coverslips were subjected to a shear stress up to 11.2 dyn cm−2. Here, the Reynolds number was lower than 2300, confirming laminar flow conditions in the bioreactor (figure 1(f)).
To assess the biocompatibility of the bioreactor system, HUVECs were dynamically cultured for 24 h at a flow rate of 1.48 ml min−1. Cell viability was detected with a live/dead assay. As shown in figures 2(a), (b) and figure S1 is available online at stacks.iop.org/BMM/13/024101/mmedia, HUVECs cultured under static or flow conditions after 24 h retained viability (static: 97.4 ± 0.01% and bioreactor: 96.2 ± 0.015%). No significant difference in the number of viable cells was detected between static and flow conditions (figure 2(e), p > 0.05). HUVECs in both conditions were CD31-positive, indicating that the phenotype of the cells was maintained (figures 2(c), (d)).
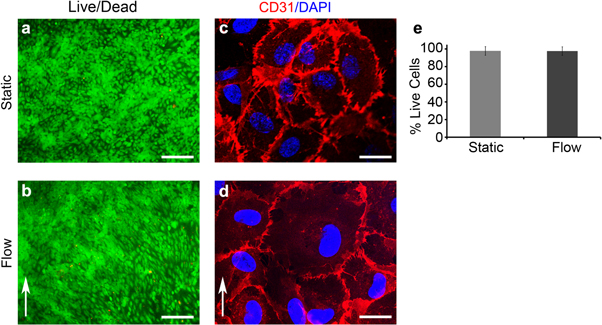
Figure 2. Fluorescence staining results 24 h after scratch assay under either static (a), (c) or dynamic (b), (d) conditions. (a), (b) Live/dead assay: living cells are stained in green and dead cells are visualized in red. Scale bars equal 100 μm. (c), (d) CD31 immunofluorescence staining (red) and DAPI (blue). Scale bars equal 20 μm. (e) Quantification of the live/dead assay from n = 3 images of both static and flow conditions. Error bars represent the standard deviation. There was no statistically significant difference between the two conditions.
Download figure:
Standard image High-resolution imageTo demonstrate functionality of the bioreactor, HUVECs were subjected to a wound healing assay (scratch assay) similar to previous studies [31] and monitored in real-time (figure 3(a)). Interestingly, HUVECs under static conditions proliferated faster (a confluent monolayer was seen within 14 h) when compared with the cells that were subjected to flow (confluence was reached after 18 h) (figure 3(b)). We observed that similar to static cultures, HUVECs cultured with exposure to a flow rate of 1.48 ml min−1 showed a round morphology and did not align (figure S2).

Figure 3. (a) Bright field images of HUVECs in the bioreactor during live-cell imaging. The black lines trace out the scratch area. The blue arrows show the scratch channels. Scale bars equal 100 μm. (b) Quantification of the reduction of scratch area over time for both static and flow conditions (each n = 3). Error bars represent the standard deviation.
Download figure:
Standard image High-resolution imageWe performed 2P-FLIM to analyze the cells under static and flow conditions in real-time in the bioreactor. The fluorescence decay curves for NAD(P)H were the best fit to a double exponential decay model, indicating the presence of two distinctly different lifetimes for the free and protein-bound forms of this coenzyme. The lifetimes of the free NAD(P)H (τ1), and protein-bound NAD(P)H (τ2), and the relative amplitude of free NAD(P)H (α1) of HUVECs cultured under different conditions are displayed as false color-coded 2P-FLIM images in figure 4. Based on these images, histograms of α1 of HUVECs cultured under different conditions were calculated (figure 5(a)). As indicated in figures 5(a), (b), α1 and τ1 of the 24 h laminar-sheared HUVECs were decreased when compared with the initial state (flow condition, 0 h). In contrast, α1 in HUVECs cultured under static conditions showed similar lifetime distributions after 24 h of culture when compared to the initial state. An increased τ2 was detected in the 24 h laminar-sheared HUVECs when compared to the cells prior exposure to defined shear (figure 5(c)).
Figure 4. Multiphoton laser-induced autofluorescence images and false color-coded images of the relative contribution of free NAD(P)H (α1) in HUVECs cultured under static or flow conditions. Scale bar indicates 100 μm.
Download figure:
Standard image High-resolution image
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Figure 5. Histograms of (a) the relative contribution of free NAD(P)H (α1), fluorescence lifetimes of the (b) free (τ1) and (c) protein-bound (τ2) NAD(P)H in HUVECs cultured under static or flow conditions.
Download figure:
Standard image High-resolution image{kind=link}
Discussion
Bioreactor systems are widely used to mimic physiological conditions in cell and tissue cultures, and to specifically impact cellular behavior such as cell migration, proliferation, and phenotype maintenance [32]. However, many bioreactors do not permit in situ imaging and analyses [33, 34]. As a result, the sample must be removed from the bioreactor system and be prepared for further assessment. Thus, it is not possible to continuously monitor the culture and obtain data at several time points. Devices exist to support continuous low-resolution visualization of cells during flow, or high-resolution visualization of cells while stretching the substrate [35–39]. Nevertheless, high-resolution imaging of intracellular structural dynamics during shear stress applications is not readily feasible.
Here, we present a high throughput bioreactor system that is designed for real-time high-resolution imaging of cells and tissues that are exposed to fluid shear stress. To our knowledge, this is the first demonstration of combining a flow bioreactor system with 2P-FLIM. The windows at the bottom of the bioreactor allowed easy assembly onto the stage of a standard inverted light microscope system without interferences with the optical setup. The newly designed bioreactor system combines four independent flow chambers that allow the assessment of distinct culture conditions simultaneously and thereby largely improves the culture efficiency. The simulation of shear stress indicated that the bioreactor provides homogeneous and reproducible laminar flow conditions with flow rates ranging from 1.48 ml min−1 to 47 ml s−1. The bioreactor system can be used to mimic both low and high shear stresses up to approximately 11.2 dyn cm−2. The system was tested using HUVECs, which are directly exposed to blood flow shear stresses in vivo. Cytotoxicity tests demonstrated that HUVECs retained a high viability after they were exposed to 1.48 ml min−1 flow within the bioreactor system for 24 h. The HUVECs showed a round morphology and no alignment occurred after the dynamic culture. This is in accordance with a previous study, in which Chiu et al observed similar characteristics of HUVECs after exposure to a defined shear stress of approximately 0.5 dyn cm−2 [40]. In this study, HUVECs were round in shape with random actin filaments located mainly at the periphery of the cells [40]. In contrast, when exposing HUVECs to a relatively high shear stress (20 dyn cm−2), the cells showed an elongated morphology and alignment with the direction of flow [40].
Here, the migration behavior of HUVECs was assessed under static and continuous flow conditions employing a scratch assay in real-time within the bioreactor system. The influence of high and disturbed shear stress on HUVEC migration has been controversially discussed as endothelial migration responses vary depending on the duration and type of applied shear stress [1, 41, 42]. For example, when HUVECs were preconditioned with a relatively high stress of 15 dyn cm−2 and then scratched, the migration was inhibited when compared with the static controls [1]. In contrast, when HUVECs were scratched without preconditioning and then cultured with approximately the same conditions (17 dyn cm−2), the migration was increased compared with the static controls [40, 41]. In our study, we observed a low shear stress-induced reduction in migration of HUVECs when compared with the static conditions.
2P-FLIM was utilized to investigate the metabolic activity of HUVECs cultured under low shear stress conditions with the aim to show the suitability of the bioreactor for real-time high-resolution imaging. Kim et al have shown that shear stress can alter metabolism in endothelial cells from glycolysis to oxidative phosphorylation-dependent mechanisms [43]. Our data is consistent with these findings as we detected decreased α1 in 24 h laminar-sheared HUVECs when compared with the static controls or with cells before expose to shear (t = 0). This indicates a metabolic switch from glycolysis to oxidative phosphorylation [44]. One possible explanation for this metabolic switch is that laminar shear stress induces apoptosis of endothelial cells by reducing glucose uptake (glycolysis) [45]. A relation between metabolic phenotypes and fluorescence lifetimes of NAD(P)H has been previously reported [44, 46]. We also observed an increased τ2 that may correlate to the elevated level of oxidative phosphorylation. Others have suggested that τ1 is associated with the cytosolic acidification in cells [47]. Interestingly, a previous study has shown that laminar shear stress induces intracellular acidification in endothelial cells [48]. The pH value changes may act as a signaling mechanism for flow-induced changes in endothelial cell metabolism [48]. Therefore, it is possible that the metabolic shifts in laminar-sheared HUVECs are regulated via pH value changes.
In our study, we showed that our newly designed bioreactor system can control shear stress stimulation parameters while allowing visualization and quantification of cell migration and cell–cell interaction under shear stress-stimulated conditions. As this bioreactor system has been designed to also house 3D substrates, such as cell-populated electrospun scaffolds or hydrogels, a future application of this bioreactor system is to study cell-biomaterial interactions. Recently, it was shown that the combination of electrospun scaffolds with shear stress resulted in the increased maturation of induced-pluripotent stem cells-derived smooth muscle cells [49]. With the here presented next generation bioreactor system, simultaneous real-time monitoring and contact-free cell state analyses are now additionally possible. Moreover, due to the combination of bioreactor technology and imaging, the system can also be used in the future for direct cell manipulation such as optoporation and optical reprogramming with the ability to quantify expression, subcellular location and trafficking of receptors in living cells as seen in previous studies [50, 51]. Other potential applications of this bioreactor system have broad implications especially with the recent advances in gene editing. For example, fluorescence-reporter genes could be monitored in vitro using this bioreactor system. This real-time monitoring would help uncover mechanisms of disease in order to improve treatments and gain mechanistic knowledge. The ability to modulate the physical parameters such as shear stress and oxygen content could play an important role in cardiovascular tissue engineering and coagulation studies. Real-time monitoring of cells could give insight into the progression and mechanism of disease, which is vital for the advancement of treatment and knowledge in the field of tissue engineering and biomaterials design.
Conclusion
The newly designed flow bioreactor system presented in this study enables the exposure of cells and 3D tissues to shear stress while allowing simultaneous real-time contact-free analyses. We demonstrated that the system is capable of providing reproducible laminar flow in a wide flow range. It is biocompatible, easy-to-use and compatible with a wide range of microscope systems. To prove the functionality of the bioreactor system, we cultured HUVECs under low shear stress conditions for 24 h. Future studies will focus on a systematic study of a long-term culture of HUVECs under various types and magnitudes of shear stresses. Future possibilities for this bioreactor range from observing in vitro cell development, including but not limited to direct cell reprogramming or stem cell differentiation, and real-time monitoring of cellular (micro)environments, cell-biomaterial interactions or cellular metabolic changes due to drug applications.
Acknowledgments
The authors thank Pirmin Lakner, Katarina Klett and Simone Liebscher (University Women's Hospital Tübingen) for their support with imaging and cell cultures. This work was funded by the Fulbright US Student Program (JAR), RISE DAAD (ZD), the Ministry of Science, Research and the Arts of Baden-Württemberg (33-729.55-3/214 and SI-BW 01222-91 to KS-L), and the Deutsche Forschungsgemeinschaft (INST 2388/30-1, INST 2388/34-1, SCHE 701/7-1, SCHE 701/10-1 to KS-L).
Disclosure statement
No competing financial interests exist.